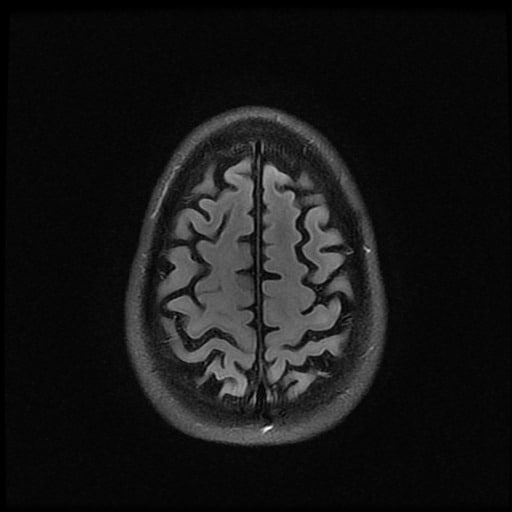
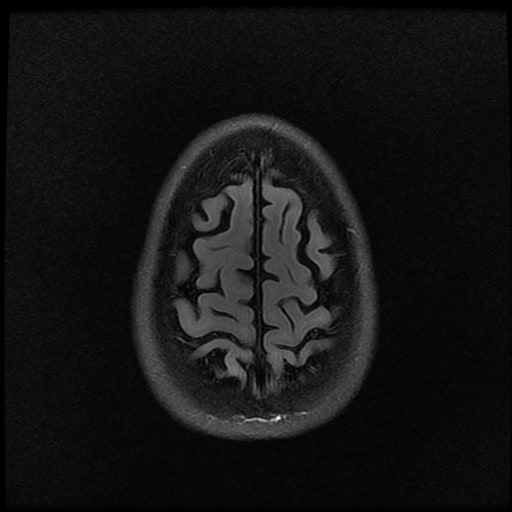
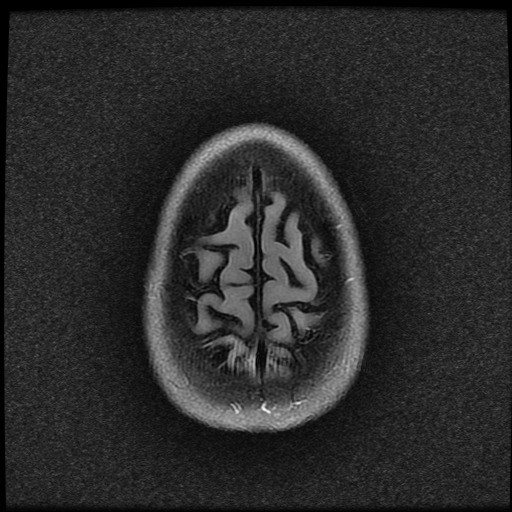
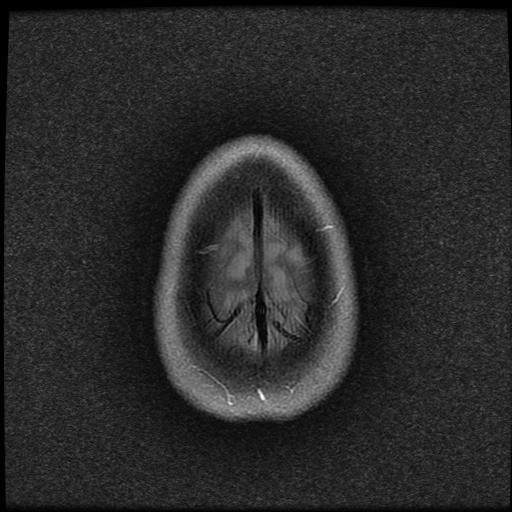
ISCHEMICKÉ CMP / KLASIFIKACE A ETIOPATOGENEZE
MELAS
Syndrom mitochondriální encefalomyopatie, laktátové acidózy a „stroke-like“ epizod
Vloženo 02.02.2020 , poslední aktualizace 31.07.2021
- multisystémové mitochondriální onemocnění
- manifestace na konci první životní dekády (široké rozmezí 2 -40 let)
- popsáno několik bodových mutací asociovaných s MELAS
- v 80 % způsoben mutací 3243A>G v genu MTTL1 v mitochondriální DNA kódující tRNALeu.
- většina případů vykazuje maternální dědičnost, avšak zřídka lze nalézt u více než 1 člena rodiny plně rozvinutý MELAS
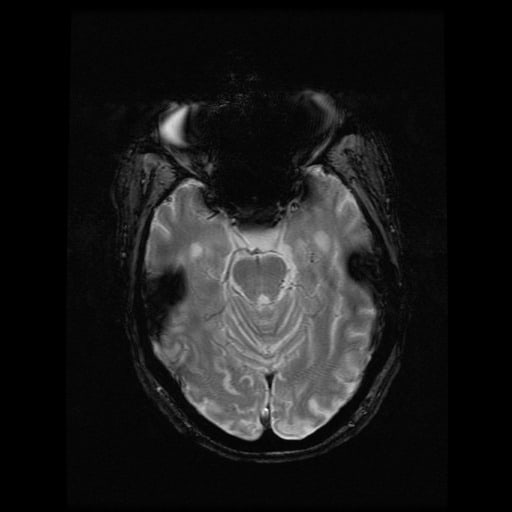
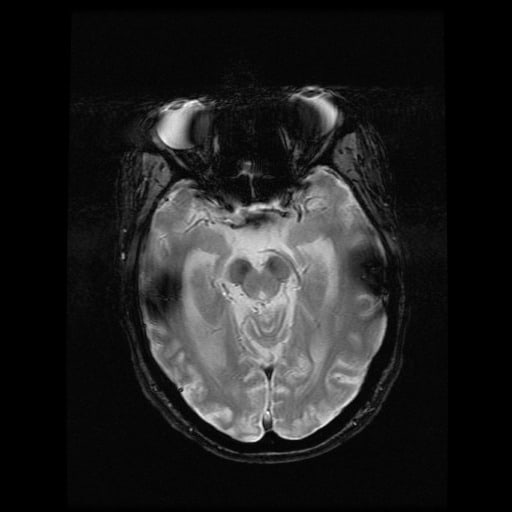
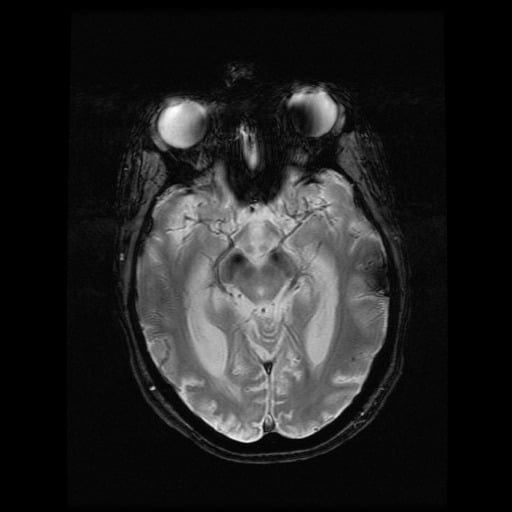
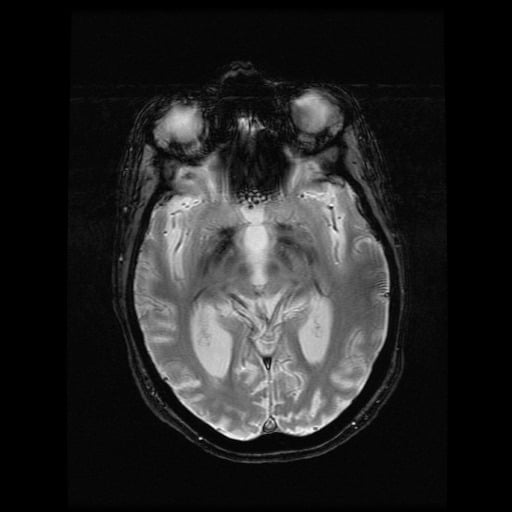
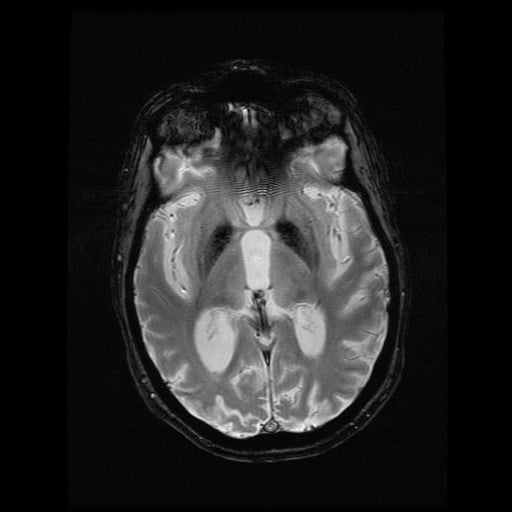
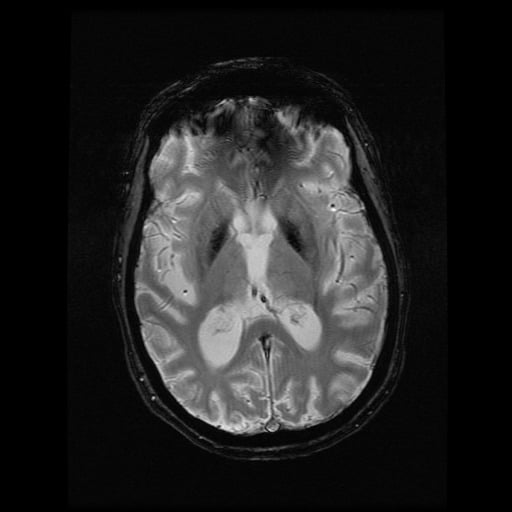
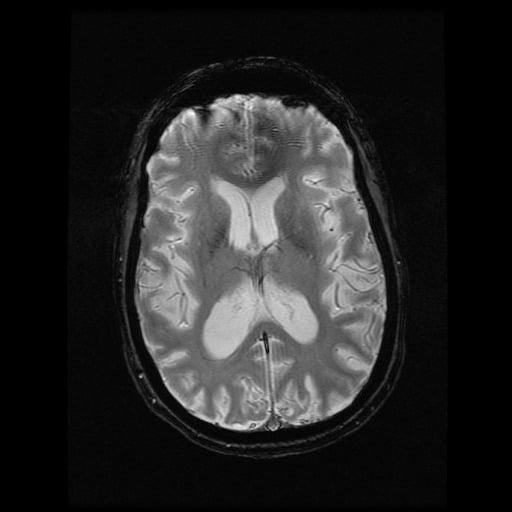
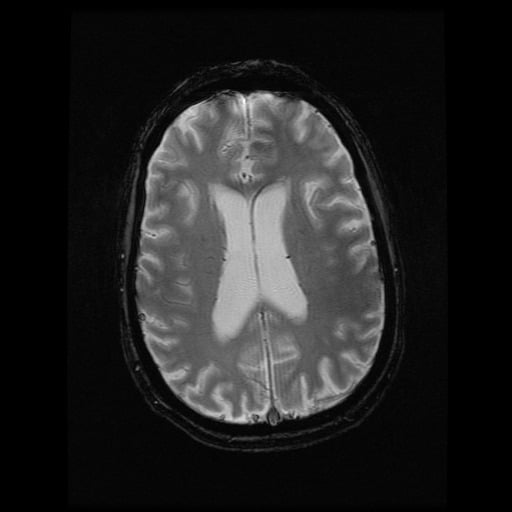
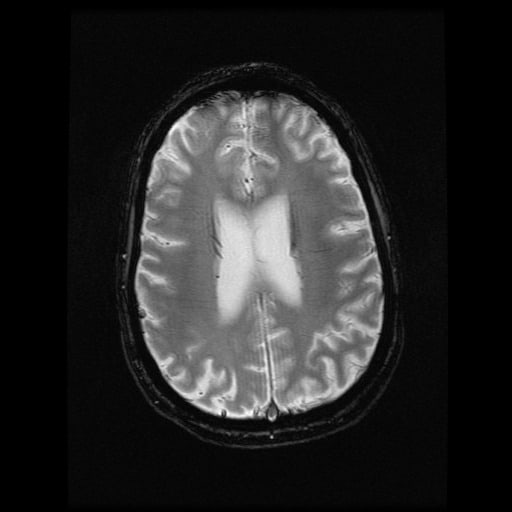
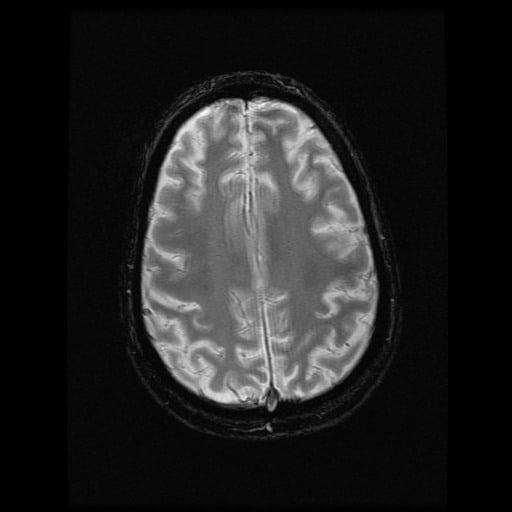
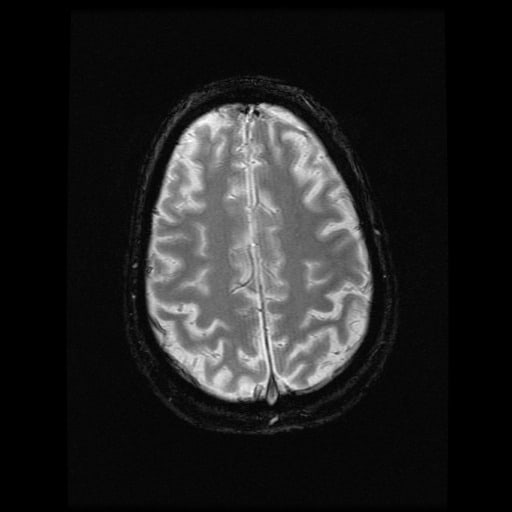
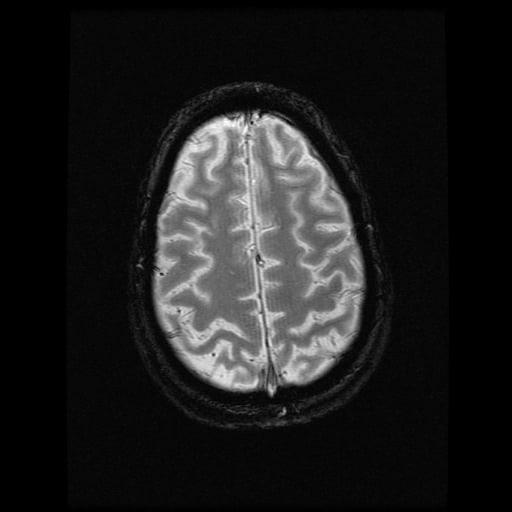
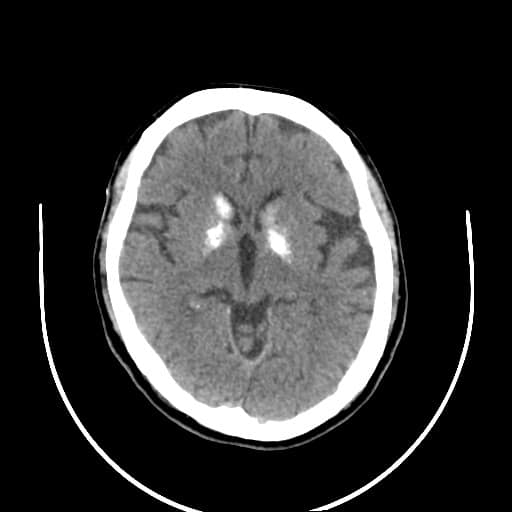
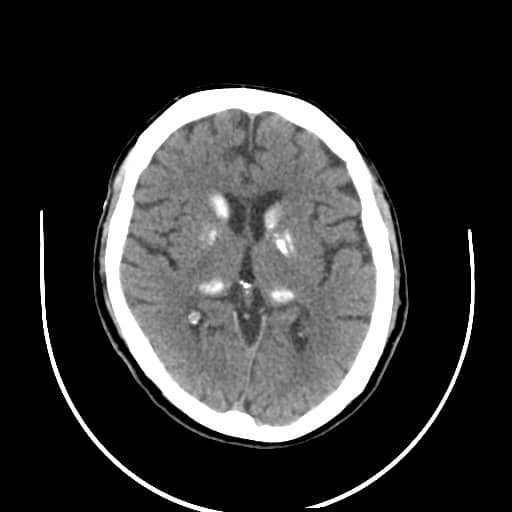
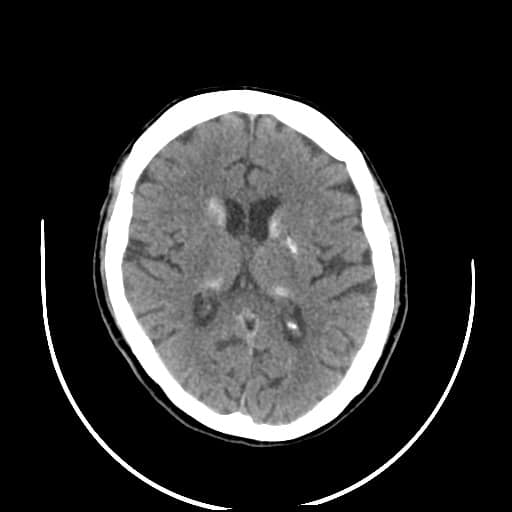
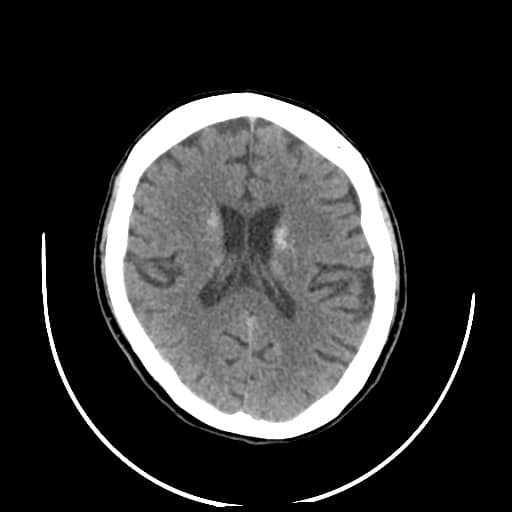
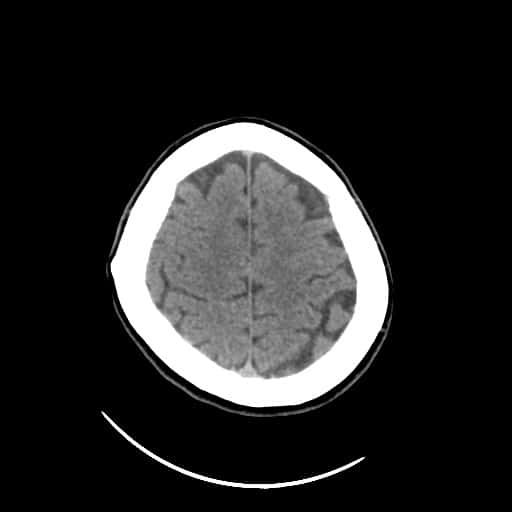
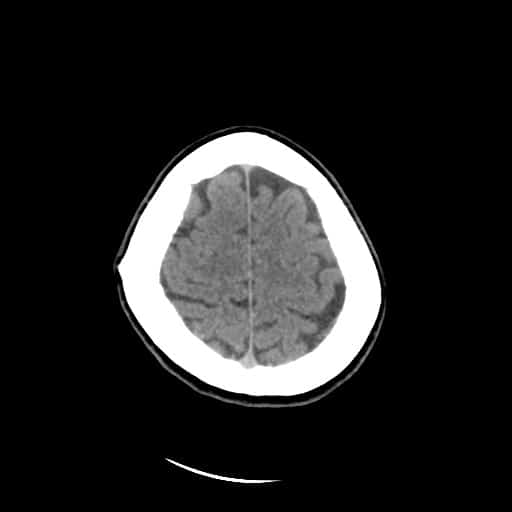
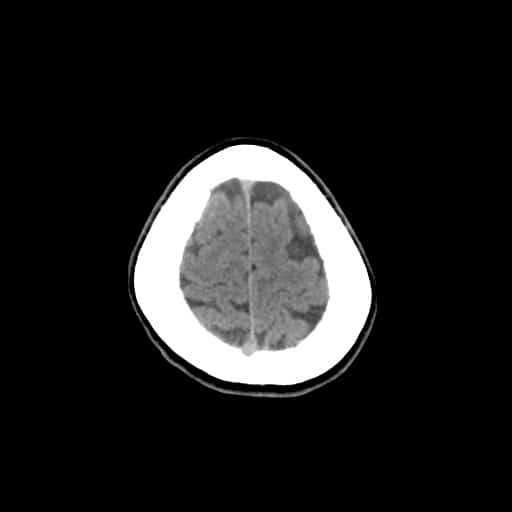
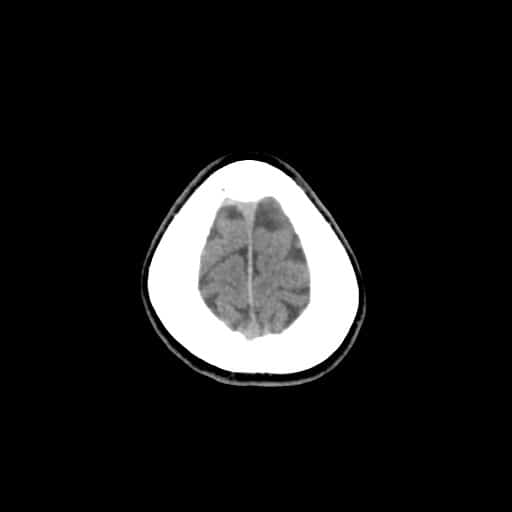
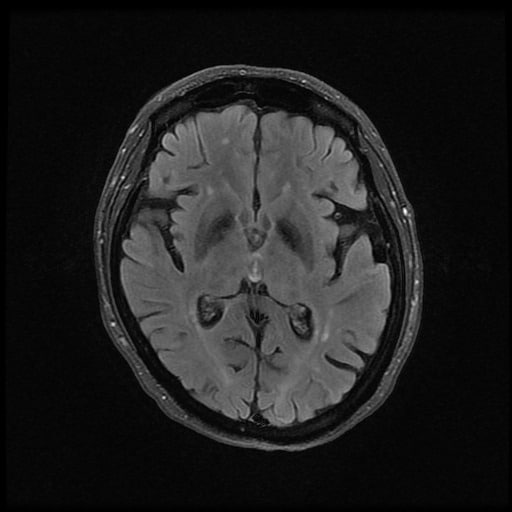
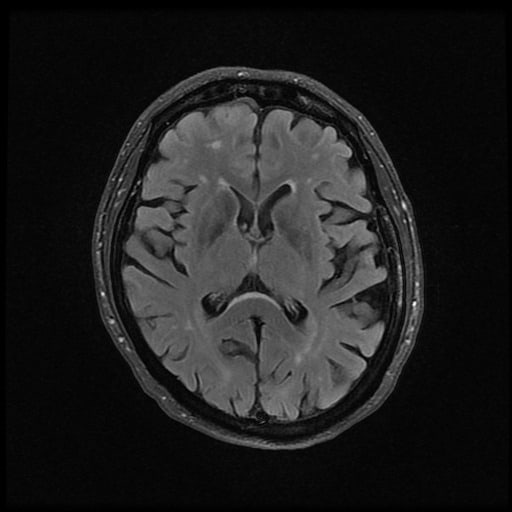
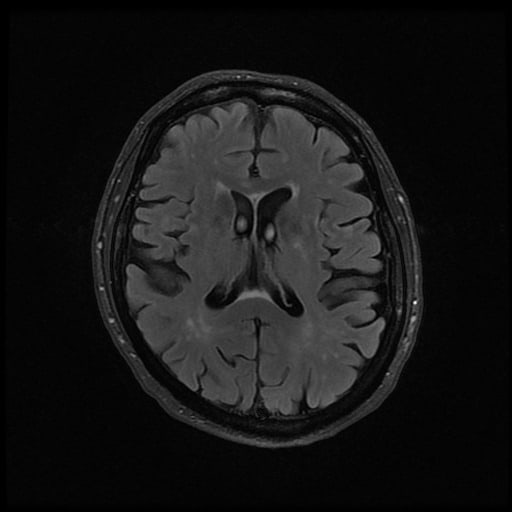
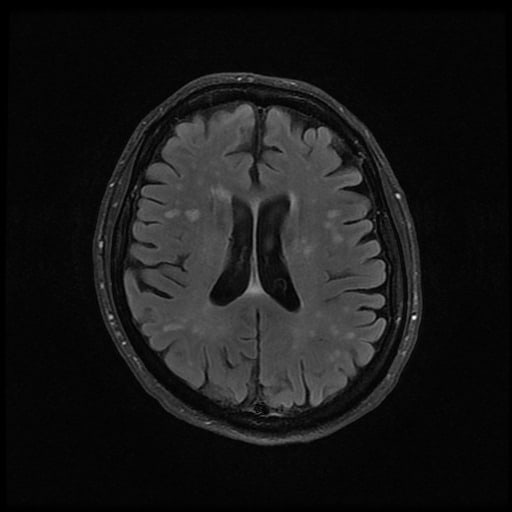
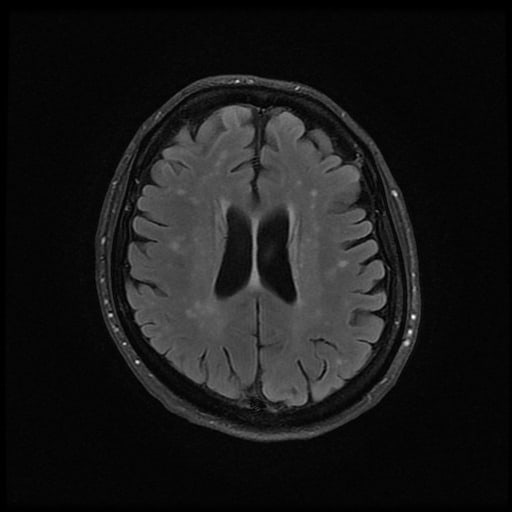
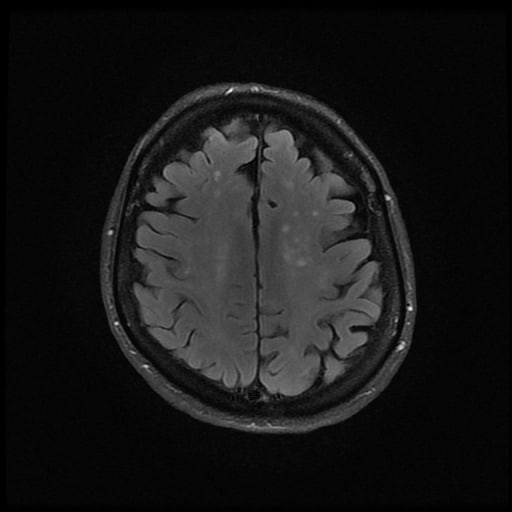
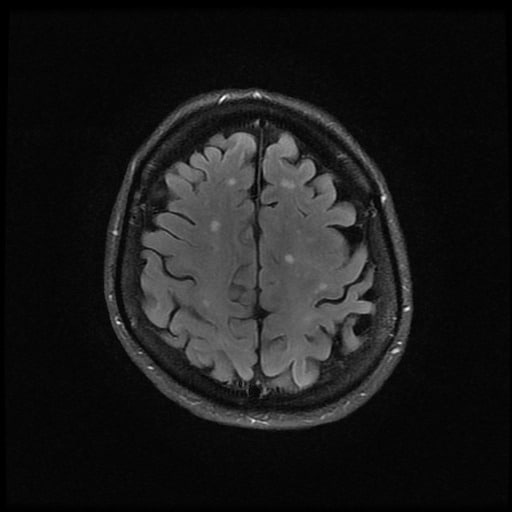
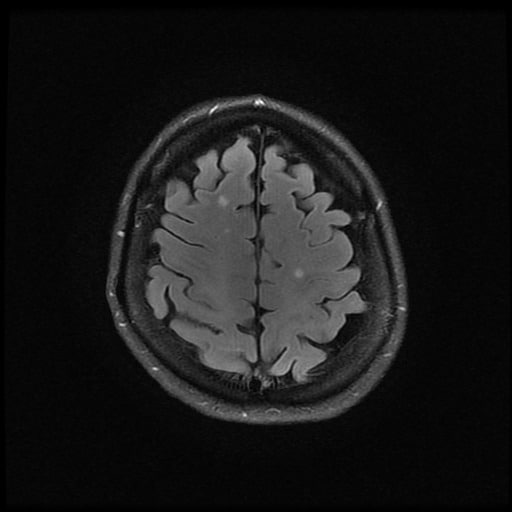
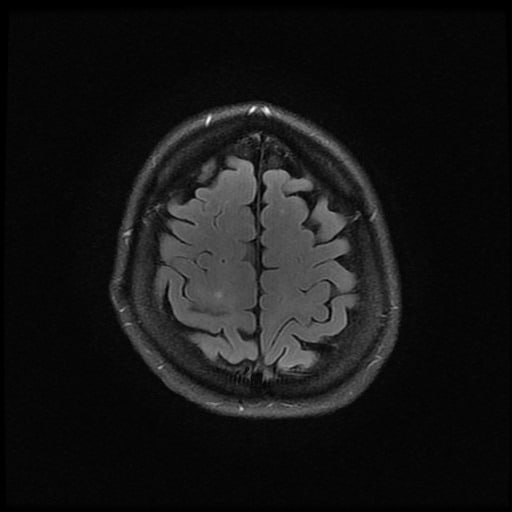
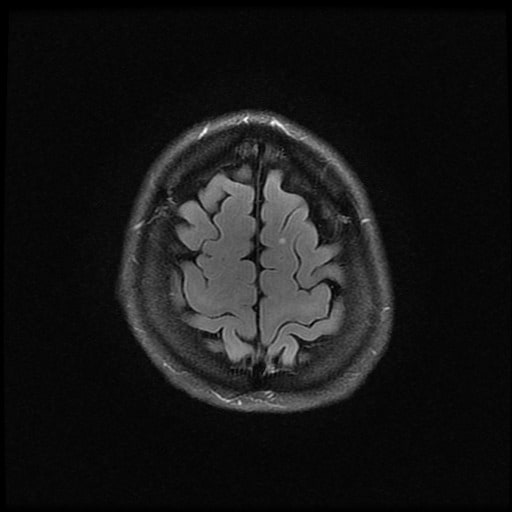
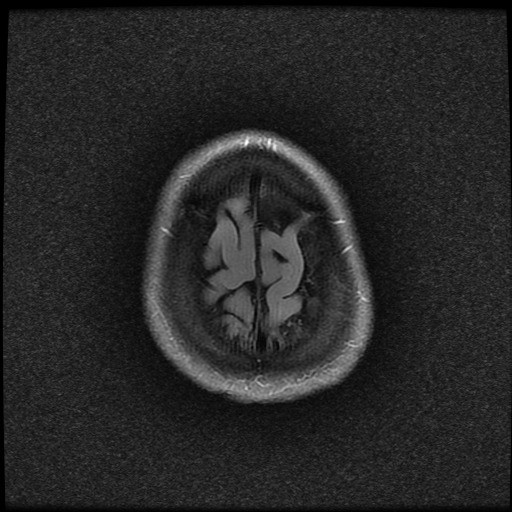
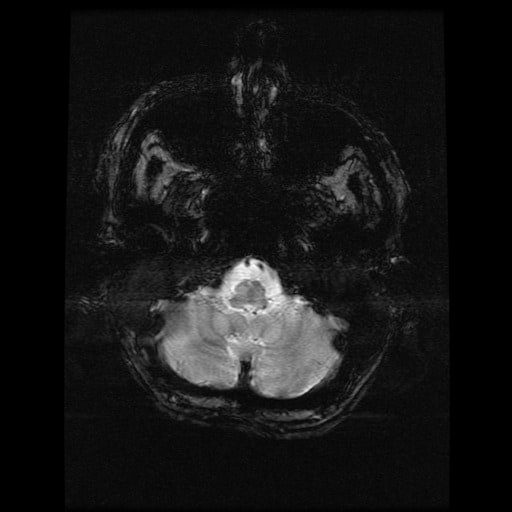
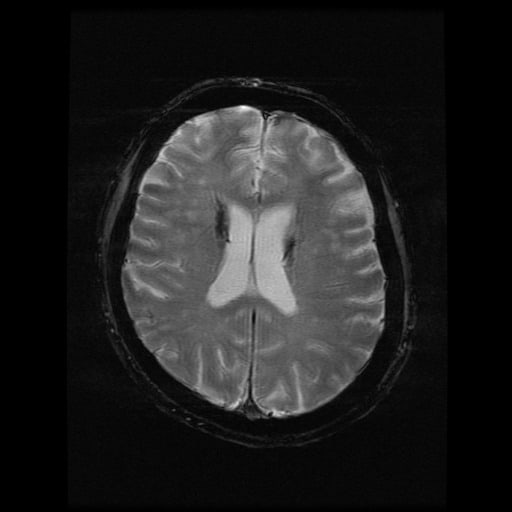
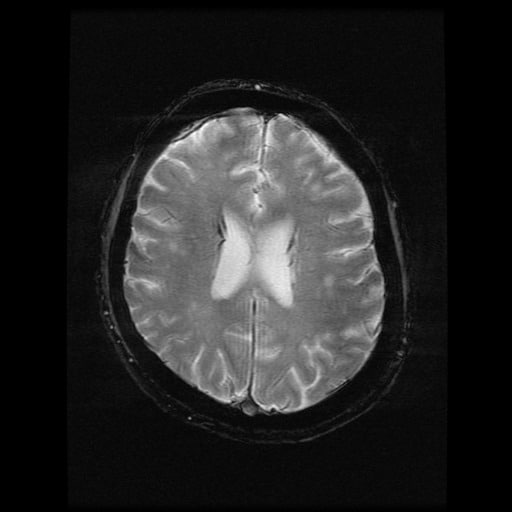
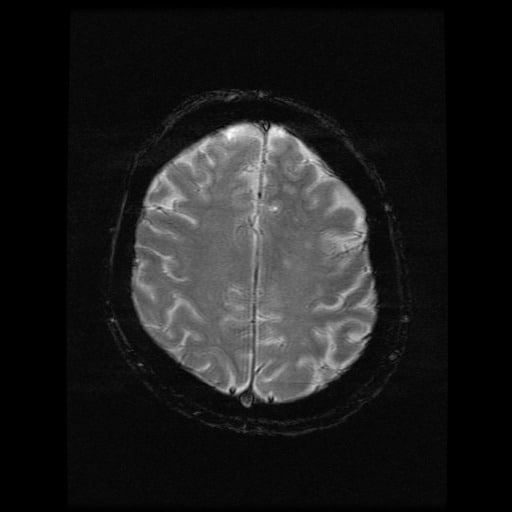
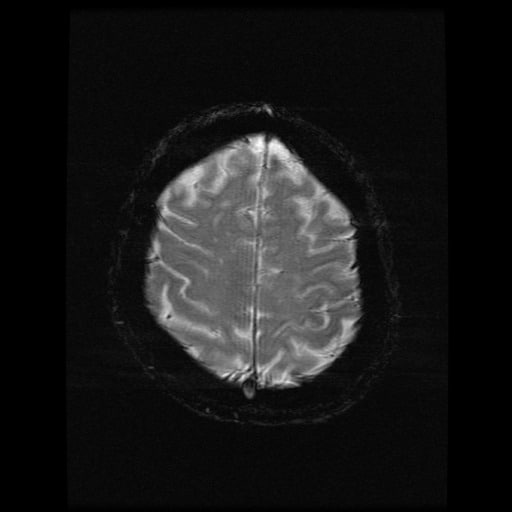
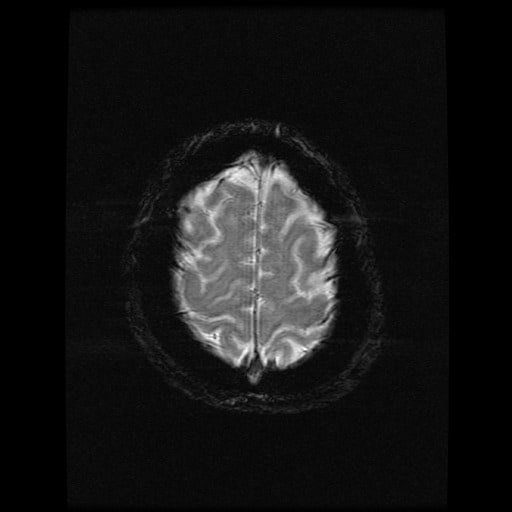
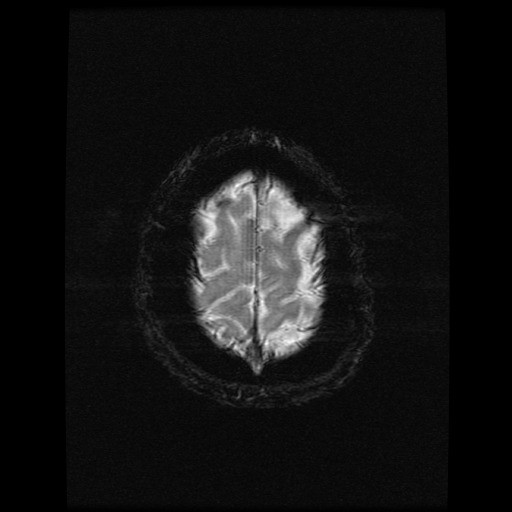
- histopatologické vyšetření mozku prokazuje spongioformní encefalopatii, charakterizovanou nekrózou, ztrátou neuronů, gliózou a mikrocystickými formacemi (status spongiosus) a predilekčně postihující kortex
- téměř v polovině případů bývají přítomny kalcifikace v bazálních gangliích
Klinický obraz
Pro potvrzení diagnózy je vyžadována přítomnost alespoň dvou ze tří pomocných kritérií:
Onemocnění se obvykle manifestuje na konci 1.dekád (rozmezí od 2 do 40 let) |
Myopatie
- svalová slabost včetně oftalmoplegie charakteru progresivní zevní oftalmoplegie (PEO)

- intolerance svalové zátěže
Systémové příznaky
- malá postava
- pigmentová degenerace sítnice
- kardiomyopatie
- diabetes mellitus
- hirsutismus
- nefropatie
Diferenciální diagnostika
- jiné příčiny cévní mozkové příhody u mladého jedince do 40 let věku
- jiné mitochondriální poruchy
- MERFF
- Kearn-Sayre
- vaskulitidy
- CADASIL (subkortikální léze a T laloky)
- MELAS je často diagnostikován jako migréna, zejména bazilární, hemiplegická či s prolongovanou aurou
Obsah dostupný pouze pro přihlášené předplatitele.
Terapie
- účinná léčba není obecně známa
- popisován částečný efekt koenzymu Q10, riboflavinu
- slibné výsledky ukazuje L-arginin 4x1g/d [Koga, 2006]
- dávkování se upravuje dle hladin argininu (ref. interval 45-130 umol/l)
- orální L-arginin prodlužuje intervaly mezi stroke-like atakami (AHA/ASA 2021)
- infúzní podání může mít efekt u status epilepticus, bolestí hlavy, poruchy vbědomí a visu (ARDEAELYTOSOL 21% + 500mg Glu 5% 20 ml/h) [Toribe, 2007]
- epileptické záchvaty reagují příznivě na antiepileptika (CAVE valproátová encefalopatie)
- část postižených matek je neplodných
Prognóza
- jde o progredující onemocnění, ke zhoršení dochází po „stroke-like“ epizodách
- prognóza je variabilní, ke smrti může dojít již v prvních životních dekádách, zejména při časné klinické manifestaci
- příčinou smrti bývá kardiopulmonální selhání či status epilepticus